KCNT2, (also known asSLICK for “sequence like an intermediate conductance K+ or KNa1.2) encodes a pore-forming potassium channel subunit which are expressed in the brain, particularly in the sub-plate of the cerebral cortex in utero and in the hippocampus and cortex in the adult. The channel is understood to have an ongoing role in the regulation of cerebral function. KNa1.2 subunits are postulated to hetero-tetramerize with KNa1.1 subunits (encoded by KCNT1 or SLICK) in at least some brain regions. KNa1.2 containing channels however have distinctive properties from KNa1.1 containing channels: they have fast activation kinetics and are sensitive to intracellular chloride levels and cell volume variations. They also have limited sodium permeability.
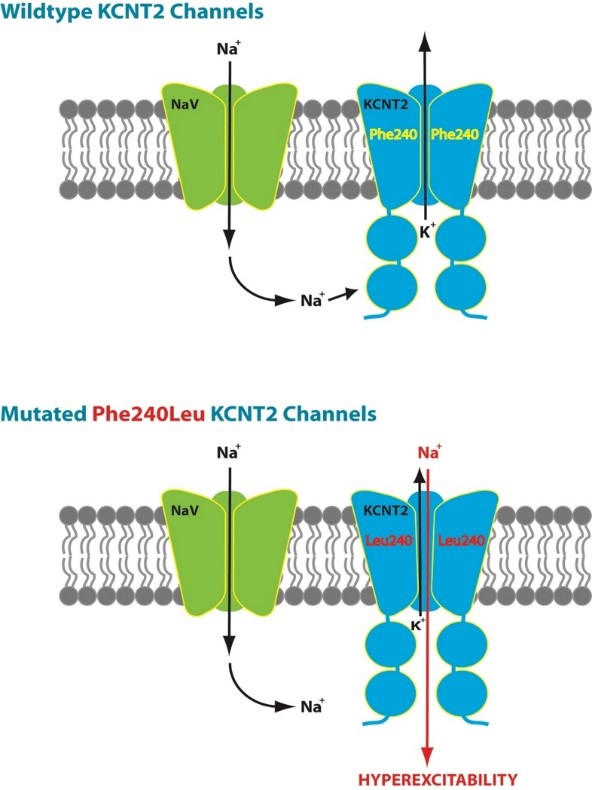
The KCNT2 variant described by our group (Gururaj et al. 2017) resulted in the amino acid substitution p.Phe240Leu. The affected residue is situated in the channel pore helix between transmembrane domains S5 and S6 (Figure 1) and had been previously demonstrated to be critical to normal KNa1.2 channel gating (Suzuki et al. 2016) Electrophysiological modelling of the mutated channel showed the mutation resulted in a dramatic change of function, essentially converting the wild-type potassium channel upregulated by chloride to a mutant channel which was no longer selective to potassium, but likely permeable to sodium, and which was downregulated by chloride (Figure 2). This is likely to result in an increased inward sodium current, disrupting the balance between Na and K exchange which is required for normal neuronal excitability.
The mutation also resulted in reduced membrane expression in a cellular model (Xenopus laevis oocytes), consistent with reduced membrane trafficking or more rapid protein degradation. Expression of the mutant channel in embryonic rat dorsal root ganglion neurons resulted in severe neuronal toxicity, and in the few surviving neurons there was evidence of membrane hyperexcitability, demonstrating a plausible pathophysiological link to the epilepsy phenotype in the proband.
The two affected children described by Ambrosino and colleagues each had a rare missense variant affecting the same amino acid residue, arginine at position 190 in the protein (p.Arg190His and p.Arg190Pro). This amino acid residue lies in the intracellular S4-S5 linker region (Figure 1) . Functional studies in HEK-293 cells showed both variants resulted in larger outwardly rectifying currents with a hyperpolarizing shift in V1/2, consistent with a gainof channel function effect.
Figure 1: position of altered amino acids in the three patients reported to have KCNT2-related encephalopathy to date.
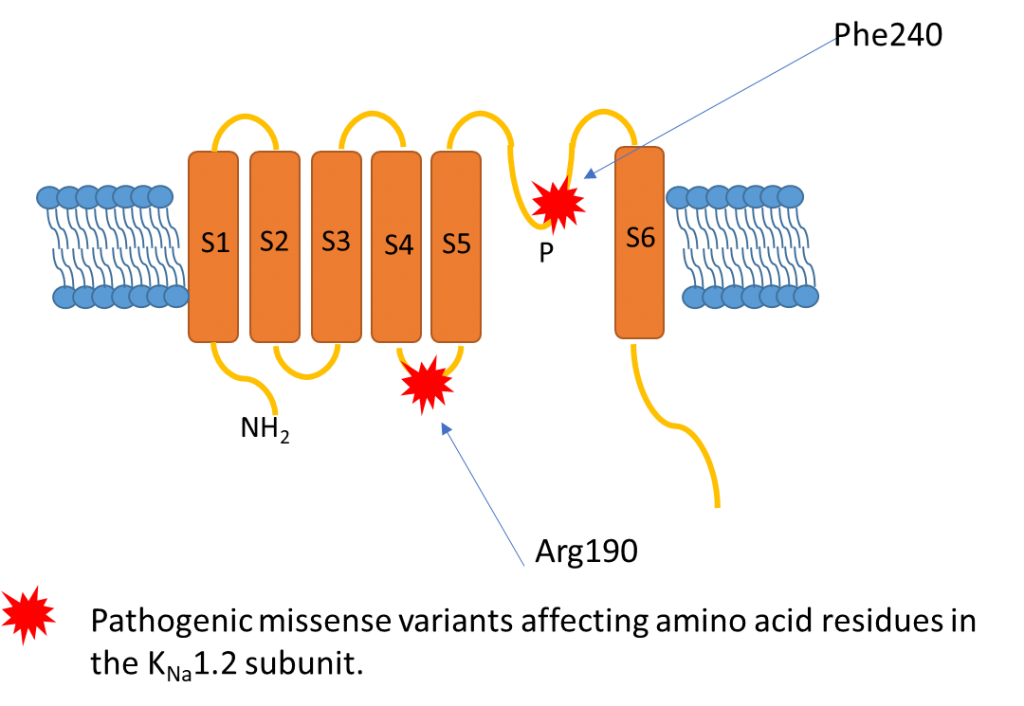
Figure 2 (from Gururaj et al. 2017): functional effect of the Phe240Leu variant in the patient described by Gururaj and colleagues.