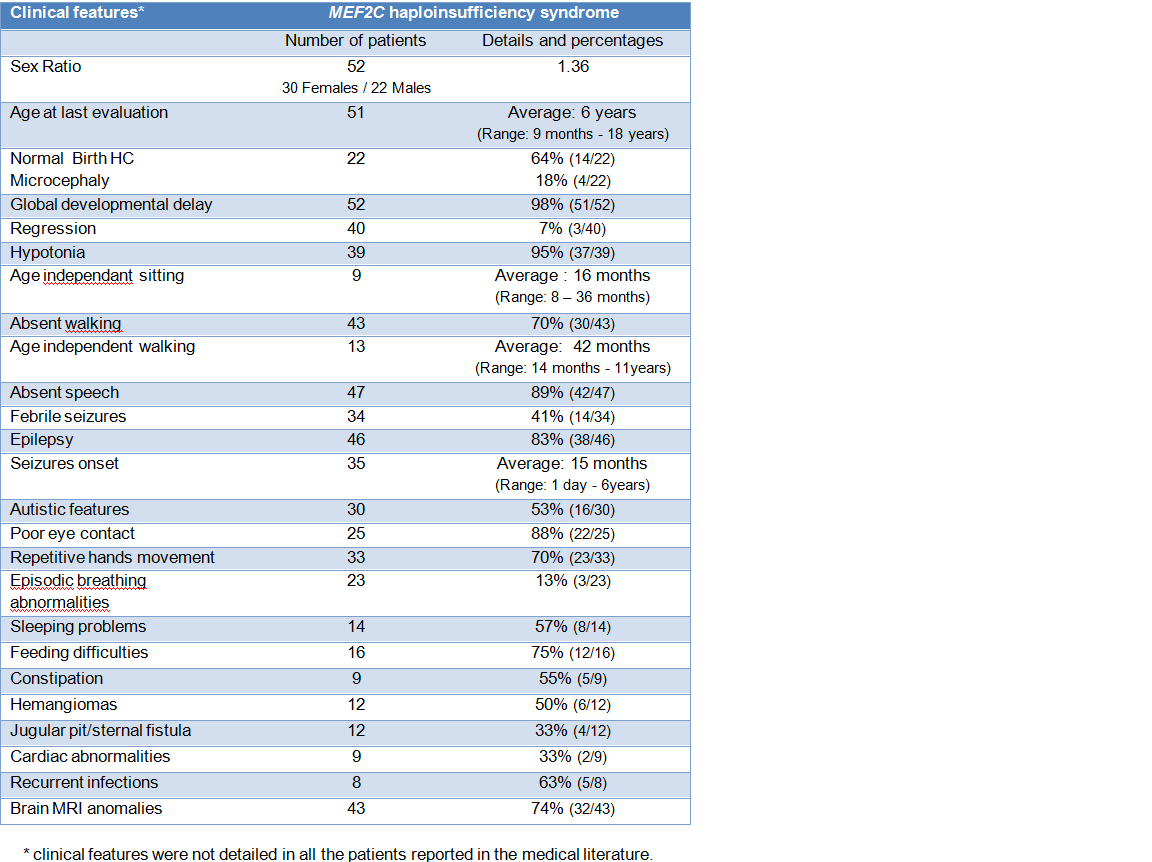
MEF2C haploinsufficiency causes an intellectual disability syndrome characterized by severe global developmental delay with absence of speech (98%), limited ambulation (70%), seizures (83%) and stereotypic hand movements (70%).
Pregnancy and neonatal course
Pregnancy is typically uneventful, but early hypotonia (95%), feeding difficulties (75%) and poor eye contact (88%) are frequent during the neonatal period.
Development
Patients manifest severe global developmental delay (98%). All patients manifest delay in gross motor development with later age for sitting, crawling and pulling to stand.
The average age for independent sitting is 16 months (ranging from 8 to 36 months) and the average age for independent walking is 42 months (14 months to 11 years).
Expressive language is typically severely impaired or absent (89%). Few patients developed some limited speech and communication abilities. Growth is generally normal.
Phenotypic overlap with Rett and Angelman syndromes has been noted. In contrast to patients with Rett syndrome, a period of developmental regression or microcephaly are rarely reported.
Distinctive morphological features
Patients are not overtly dysmorphic, but do present with distinct minor facial characteristics, including a high and wide forehead, flat nasal bridge, epicanthic folds, tented upper lip, down turned corners of the mouth, short columella and a short philtrum with prominent pillars.
Some specific craniofacial malformations were previously reported in the medical literature. Two patients had ‘question mark ears’ (Gordon et al. 2017) and one patient had a cleft palate (Vrecar et al. 2017).
Two patients had iris coloboma (Cardoso et al. 2009, Sobreira et al. 2009).
Presence of hemangiomas and fossa abnormalities (pit or sternal fistula) are recurrent findings in patients with MEF2C haploinsufficiency syndrome (50% and 33% respectively). Not all individuals with MEF2C pathogenic variant have hemangiomas and it may depend on the pathogenic variant (see molecular characteristics).
Epilepsy
Most of the patients have variable types of epilepsy (83%), although they can also be seizure-free. Seizures arise typically during early childhood or infancy. The average age for seizure onset is 15 months (1 day to 6 years). Most of the time, the seizures respond well to medication. Febrile seizures are also common (41%).
Behaviour
Most of patients show stereotypic behaviour with repetitive movement (70%), particularly hand flapping, clapping, mouthing, head rocking and hand biting.
Autistic features were reported in 53% patients.
Brain nomalies (74%)
Minor brain anomalies have been described with delayed myelinisation and cortical malformations. Brain scans showed mainly cortical atrophy and moderate ventriculomegaly, abnormalities of corpus callosum and posterior white matter abnormalities.
Cardiac anomalies (33%)
One patient had concentric myocardial hypertrophy (Engels et al. 2009) and one patient had patent ductus arteriosus (PDA) closed with a coil and patent foramen ovale (PFO) (Vrečar et al. 2017).
Other symptoms
Commonly reported symptoms are tendency to recurrent infections (63%), constipation (55%) and episodic hyperventilation (13%).
Some patients also had ametropia (myopia, hyperopia) and strabismus.
Clinical characteristics of patients with MEF2C duplication
Three patients had global developmental delay. The oldest patient had impaired but comprehensible speech (Le Meur et al. 2009). Another patient had seizures from 2 years of age.
Termination of pregnancy of a monochorionic twin pregnancy, with corpus callosum anomalies and prenatal identification of MEF2C duplication, was recently reported (Cesaretti et al. 2016).
More patient data is needed to examine the clinical presentation of patients with MEF2C duplication in more detail.