The USP7 gene is located at chromosome 16p13.2, and encodes a deubiquitinating proteolytic enzyme that can cleave multiple ubiquitin chain linkages. Previously, USP7 was shown to regulate ubiquitination of a variety of proteins with varying functional diversity. Indeed, USP7 has been implicated in transcription, DNA repair, immune response, and cancer, including the MDM2-p53 mediated pathway.
USP7 (Ubiquitin-Specific Protease 7) was reported to function in the endosomal protein recycling pathway via its incorporation into the MAGEL2-USP7-TRIM27 (MUST) complex. USP7 is a component of MUST which uses ubiquitin to activate WASH, which adds actin to the endosome, which instructs the endosome to send important proteins back to the surface of the cell where they belong.
Interestingly, USP7 actually has two functions on WASH: it does not only promote ubiquitination of the actin nucleation promoting factor WASH by preventing TRIM27 autoubiquitination and subsequent degradation, but it simultaneously limits WASH ubiquitination through its direct deubiquitination (Figure 1).
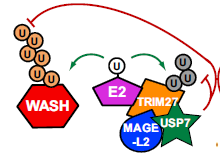
Figure 1. Model for dual activities of USP7 in preventing TRIM27 from auto-ubiquitination-induced degradation, but also limiting WASH ubiquitination levels and activity (Hao et al. 2015).
USP7’s dual function helps with precision control and fine-tuning of the protein recycling process. When the USP7 gene has a mutation, this process doesn’t work, and the recycling and degradation of proteins in the cell will be changed, causing the symptoms seen in USP7-related diseases.
Mutations in USP7 are diagnosed through either whole exome sequencing or chromosome microarray analysis (CMA), and can be either point mutations or gene deletions (Figure 2). Fountain et al. (2019) analyzed a cohort of 23 patients with molecularly confirmed de novo heterozygous deletions/variants involving USP7. Among these patients they found individuals with deletions of chromosome 16 including all or part of USP7, individuals with nonsense, missense and splice-site variants in USP7.
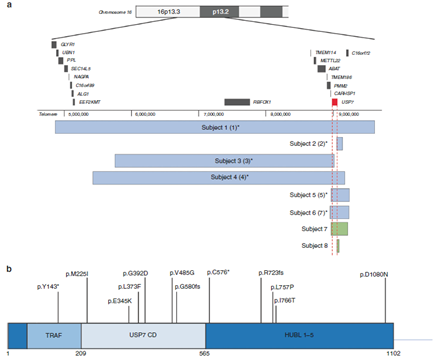
Figure 2. USP7deletions and point variants. (a) USP7 whole and partial gene deletions in eight individuals, identified via CMA or genome sequencing. Genomic coordinates based on the Human Genome Assembly hg19. (b) Nonsense variants, frameshifting indels, and missense variants in USP7 Fountain et al. (2019). The consistency of clinical features among all individuals, regardless of variant type, supports the pathogenicity of these variants and haploinsufficiency as a pathomechanism for USP7-related disorders.
The consistency of clinical features among all individuals, regardless of variant type, supports the pathogenicity of these variants and haploinsufficiency as a pathomechanism for USP7-related disorders.