GABA-Transaminase (GABA-T) is the initial key enzyme involved in GABA degradation. Deficiency of this enzyme is inherited in an autosomal recessive pattern and was initially reported in 1984, with two cases reported in the same family suggesting mortality within the first two years of life. Only a small number of affected individuals have been published, although more cases are being identified with increasing use of phenotype specific gene panels and whole exome sequencing. The initial reports described neonatal or early infantile onset encephalopathy, although other phenotypes are emerging with increased recognition. Recent reports indicate survival into adolescence and adulthood.
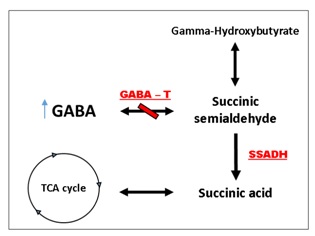
Figure 1: GABA degradation pathway. GABA is normally converted via GABA-transaminase into succinate semialdehyde, which is then broken down to succinic acid by succinate semialdehyde dehydrogenase (SSADH). The absence of GABA-T leads to increased endogenous GABA in the brain.
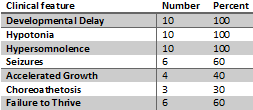
GABA-T deficiency is a multisystem disorder characterized by developmental delay, hypotonia, hypersomnolence, epilepsy, and choreoathetosis. Accelerated linear growth and failure to thrive are also characteristics. The accelerated linear growth has been attributed to growth hormone promoting effects of GABA. Children with GABA-transaminase deficiency have profoundly impaired development. The majority do not achieve normal developmental milestones of infancy, such as sitting unassisted.
Table 1: Clinical Manifestations GABA- T (N=10) (Koenig et al. 2017)