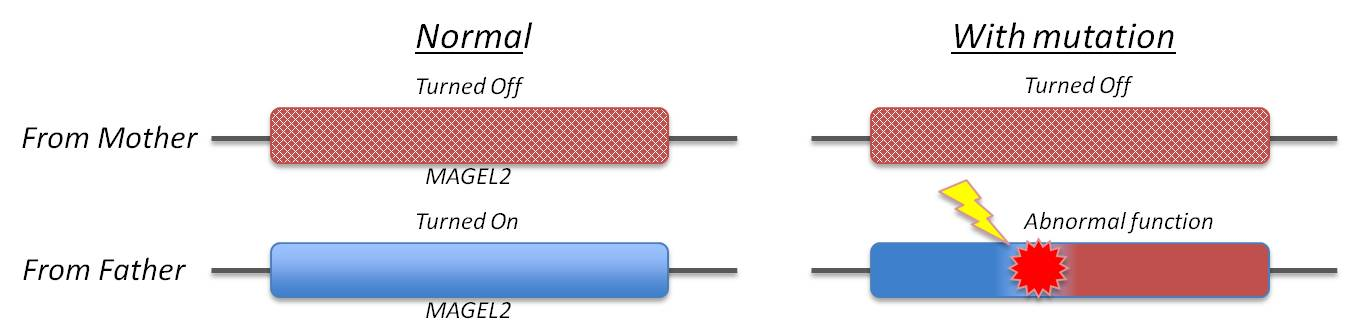
Schaaf-Yang syndrome is molecularly defined by the presence of pathogenic truncating mutations of MAGEL2 on the paternal allele of the affected individuals.
Molecular confirmation is necessary for a definitive diagnosis, though a familial pattern of inheritance coupled with the manifestation of specific Schaaf-Yang syndrome phenotypic characteristics can indicate this diagnosis.
To date, over 40 individuals with truncating mutations in MAGEL2 have been identified. As well, three cases of individuals harboring an in-frame deletion of seven amino acids (21 nucleotides) have also been identified.
Typically, individuals are identified via whole exome sequencing, with subsequent confirmation via Sanger sequencing. However, it is possible to perform Sanger sequencing only if clinical features make a diagnosis of Schaaf-Yang syndrome highly likely. Targeted Sanger sequencing of MAGEL2 can also be used to confirm the presence of the variant in question on family members, to identify the maternally imprinted, paternally expressed inheritance pattern.
To confirm the inheritance pattern in a proband, a methylation-sensitive restriction enzyme digestion can be performed to establish the parental origin analysis of alleles.
This method digests the existing un-methylated paternal allele, leaving only the (unmethylated) maternal allele, which is then amplified and sequenced. If the variant in question is still present on the sequencing, that indicates its location on the maternal allele, making it unlikely to be the cause of the observed phenotypes (and likely not Schaaf-Yang syndrome).
However, if no variant is detected after digestion, ipso facto the variant is harbored on the paternal allele and is characteristic of Schaaf-Yang syndrome.
Indeed, due to the range of phenotypes and potential severity, prenatal molecular diagnosis can be obtained in individuals suspect for harboring the pathogenic variant. Pre-implantation genetic diagnosis may be offered to affected families, in particular when the father carries a pathogenic MAGEL2 mutation (on his maternal allele).
Interestingly, patients encompassing deletions of MAGEL2, but not the SNORD116@ cluster on chromosome 15q, have been described. Their phenotypes appear milder than those with truncating MAGEL2 mutations. They have presented with hypotonia, feeding difficulties during infancy, kyphosis, and a slight delay in fine and gross motor skills.